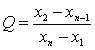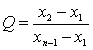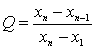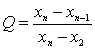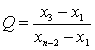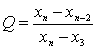�л�������Դ����
SD322��89
ȼ�ϼ��鹤��ȫ������������
�л�������Դ��1989-3-27���� 1989-10-01ʵʩ
1 ����
1.1 ���÷�Χ
���������ڵ�����ҵ�����������С���ƵȲ���ȼ�������ҹ�����ȫ������������
1.2 ����
ȼ�������ұ�����ȼ�Ϸ������ݱ��뾭��������������飬����Ҫ���ڣ�
1.2.1 �����볧ȼ�ϵ�������
1.2.2 ���۹�¯�豸���еľ����ԣ�
1.2.3 Ԥ���¯��ȫ���������
1.2.4 ����¯��Ƶ�ԭʼ���ݣ�
1.2.5 �����ڹ�¯���з�ʽ�����ݡ�
1.3 Ҫ��
1.3.1 Ϊ��֤ȼ�Ϸ�������ȷ�ɿ���ȼ�������ұ�����ѭ�����еĸ���涨�����Է�������������ʱ��������ԱӦ�ṩ����������˵���ͼ�¼��
1.3.2 ȼ�������ҵ�ȫ����������������Ӧ�ᴩ��ȼ�Ϸ����ĸ������ڣ��������һ�����ʩ�����ã�����������У��ʹ�ã�������ϵĹ�Ӧ�������������IJ�������Ʒ�ı��ܣ�����������ѡ��ֱ���������ݵĴ��������������Լ����鱨����ⶨ�ȣ��������Ա�֤���������ȷ���뾫�ܶ�Ϊǰ�ᡣ
2 ��������ʩ����������
2.1 ��������ʩ
ȼ����������ʩ��ָ���ȼ�Ϸ�������������Ļ�����ʩ����Ҫ�����������䡢��ƽ�䡢�����䡢���ȼ䡢���ȼ䡢���ؼ䡢����ϵͳ����������ϵͳ������ˮ��������ϵͳ�ȡ���Щ������ʩ���밴��һ���ı���Ҫ���졣�����ҵĻ�����Ӧ����糾�������������¡���������Ӱ�죬����Ӧ�����õ�������
2.2 ���������������
2.2.1 ������Ҫ�������ߴ�(������4��4.5m),�ɹ����ã�ǽ���࣬����⻬��Ӳ(��ˮĥʯ�̳�)�����ڽ����������������
2.2.2 �������ڲ�Ӧʹ��ú��¯�������ܹ���Ⱦ���ڻ�����ȡů�豸������ˮ��Ҫ��װ�ڸ�������ĺ���λ�ã������ʵ�����ˮ�ء�����Ҫ����ר�õ��к������ﴢ��Ͱ�������ڰ�ȫ�ص㣬��Ϊ�������Ա㼯�д�����
2.2.3 ������Ĺ̶��豸��ͨ���������̨��ҩ��������������(���)������װ�õȶ�Ӧ������Ҫ����ơ�����Ͳ��á���ͨ�����Ҫ�����л��ܼ��Ĵ�������Ʒ�����⼰һЩ�к�������ų��ȡ���ˣ�һ������Ҫ���ͨ��������䴰���ִ�ʱ��������������ӦΪ30m/min������̨�ĸ߶ȺͿ���Ҫ���У����ڲ�����һ��Ϊ900mm��750mm��̨��Ҫ������IJ����̳ɡ�
2.2.4 ������Ĺ���Ӧ���㣬����ϵͳ�������Լ���Դλ�õ�Ӧ�����ڷ�����������ɫ�жϣ��������ṩ�������ʸС�
2.2.5 ��������Ӧ��������������ģ���Ӧ���ڼ�飬��֤��ʱ�ɹ�ʹ�á�
2.3 ��ƽ�����������
2.3.1 ��ƽ��Ӧ��������ֱ�����䣬�����¶Ⱥ�ʪ��Ӧ�����ܱ����ȶ�������Ҫ�������Ҫ��͡������������(�紩�÷磬���Ȳ�����������)���Լ��ٻ�����������ƽ�������ܶȵ�Ӱ�졣��ֹ����ƽ����ʹ���κμ����豸��
2.3.2 ��ƽ�丽����������Դ����ƽ̨Ҫ���ƽ�ȣ���Ӧ����ǽ�ڣ����ü��������������̨��Ӧ���档
2.3.3 ��ƽ����ƽ̨�ϵİ�װ���Ҫ����0.5m��̨��Լ800mm��̨��Լ600mmΪ�ˣ�Ҫ���㹻�Ĺ����棬����һ�����ʵij��������ƽ�ĸ����ȡ���ƽӦ�з����֡�
2.4 ���������������
2.4.1 �������ǰ��Ѳɼ���ú�����Ƴɷ���ú���Ĺ����䡣��Ӧ���ܷ硢����Ϯ����糾����Ӱ�죬��װ���ŷ��ȵȳ����豸��
������ӦΪӲ�ʵ��棬�����ڵ����������ʵ������5mm���Ϻ�ȵĸְ塣����ú������Ӧ����Դ������ֱ�䡣
2.4.2 ��������Ӧ���������ĸ��������豸���繩����ú������ʽ�����������ϸ��ú����Բ����������Ʒ������ܷ�ʽ������ȡ�
2.4.3 ������Ӧ���и��ֹ��ı�ɸ�����õ�Ϊɸ��150��100��50��25��13��3��1mm�Ĵ�ɸ�Լ���Ϊ200��m��88��m�ı�ɸ������Ӧװ�л�е��ɸ�豸��
2.4.4 ��������Ӧ��������ú���ĸ��ֹ�����е�����������������豸����ͬ���Ķ�������ʮ�ַ����塢ƽ�����¡���������п���̻��´��̡�̨�ӡ�������ƽ��ëˢ���������綯��ɨ�豸���������ù��ߡ�
2.4.5 ������Ӧ����רΪ�����Ʒ�õĹ��ܡ�
2.4.6 ������Ա����������ʱ��Ӧ����ר�õ�������Ь��ñ�����ֺ����ף��Ա������ʱ��Ⱦú���ͱ���������Ա������������Ӧ������ϴ�ֺ�ϴ����ʩ��
2.5 ���ȼ����������
2.5.1 ���ȼ��ǹ��ⶨȼ�Ϸ�������ר�ù����䡣������Ӧ���ڱ�������ֱ��ĵط����ڲ��ȼ��ڲ��ý����������顣
2.5.2 ����Ӧ�����ܱ����ȶ���ÿ�βⶨ���±仯�Բ�����1��CΪ�ˣ��������¶��Բ�����15��30��C��ΧΪ�ˡ�
2.5.3 �ڲⶨȼ�ϵķ�����ʱ������Ӧ����ǿ��ͨ�缰��Դ���䡣
2.5.4 �����²�������2.5.2��Ҫ����Ӧ��װ�յ��豸��������Ӧ�����ʵ�λ�ã��������¶��ȶ����ɽ��з������ⶨ��
2.5.5 ���ȼ���Ӧ���ղ��ȵIJ���Ҫ�����ò��ȵIJ���̨���Ա��ڹ۲����±仯�Ͳ���Ϊ���������ָ����豸������ƿ��ѹ������ר����ƽ(����Ϊ1 g������4��5kg)��ҲӦ�����ڰ�ȫ���ֱ��ڹ����Ĺ̶�λ�á����ֹ��ߺ�������ר�ã���Ӧ������������á�
2.6 ���ȼ����������
2.6.1 ���ȼ���ָ���ø������͵ĵ����豸�������¯�������䡢ɰԡ��ˮԡ�����Ȱ塢�۵�¯��Ԫ�ط���¯�ȵ�ר�ù����䡣
2.6.2 ���ȼ�ӦΪӲ�ʵ��棬�����豸Ӧ�����ڼ�̶��Ҿ�Ե���õ�̨���ϣ�̨ǰ�Ĺ���������Ӧ���о�Ե�塣�������豸Ӧע��ӵ��ߣ����ʹ���е��ߵ����ײ�ͷ��
2.6.3 ���ȼ��ڵ�ԴӦ���㹻���������ڵ����豸������ɢ���������ȣ�����Ӧ��װǿ�����ŷ�װ�á�
2.6.4 ���ȼ���Ӧ�����ʵ�������豸(���ڵ�������)��
3 �����������豸����������
3.1 �����豸��ѡ��
���������豸�ļ��������������������Ŀ���涨������Ҫ������ȡ��ȷ�ɿ����ݵĻ�����֤�����õ������豸��Ҫ�������ռ�飬�����ܺ��Ӧ����ʹ��˵����Ĺ涨��
3.2 ������У
��������ľ��ܶ���ȷ�ȵ�Ҫ���й�ֱ�Ӽ��������Ͳ����DZ�(����ƽ���¶ȼơ�ѹ���Ƶ�)�����붨�����йط�����������У����ʹ�á����йؼ�Ӽ����ļ������(�����ȼơ�Ԫ��¯��)��Ӧ�ñ�ú��������ʽ���У�����辫ȷУ�����������ɸ�������ľ���Ҫ����������Ա���о����Լ�顣
3.3 ������ʹ�ú�ά��
3.3.1 ר����������������(�����������в�)Ӧ��ר�˱��ܣ�����������������������Ӧ��������˵���飬���պ͵��Լ�¼�������ĸ��ֳ�ʼ���������ڱ���ά�ޡ��춨��У�Լ�ʹ������ĵǼǼ�¼�ȡ�
3.3.2 ʹ��ר��������������Ա���뾭����ѵ�����˺ϸ���ܲ���������������Ա�������Ľṹ������Ӧ�г���˽⣬�⽫��������ȷʹ�������Ͷ��������ϵļ����ų���
3.3.3 �����İ�װ�����ԡ�ʹ�úͱ���ά��Ӧ�ϸ�������˵�����Ҫ��ʹ������ǰӦ�ȼ�������Ƿ���������������������ʱ��Ӧ��������ԭ���ų����Ϻ����ʹ�ã�����������ë��������Ͷ����ת��
3.3.4 �������������Ӧ��ԭ��������������(������ͷҪ�þ������)���Ǻ÷����֡�
4 ���������������������������
����������������������в�����������������ʯӢ��������������������������ȡ��Դ�������Ҫ���������Ŀ��(��洢�Լ����������������������)�����л�ѧ��Ӧ���ص��Լ���������ȷ��ѡ��ʹ�á����ʵ���ѡ���ʹ�ô�������������ᵼ�´����������¹ʡ�
4.1 ����������ѡ��У��ʹ�ú����
4.1.1 ѡ��������ʱ��Ҫ��������;ѡ��ͬԭ�ϵ��Ƴ�Ʒ����ѡ�����Һ����Ҫ���Ƚ��л�ѧ��Ӧ������ʱ��Ӧѡ��Ӳ�ʲ������Ƴ�Ʒ(�������ҺҲ��ѡ�þ���ϩƿ)��
4.1.2 ���ڶ��ݵIJ���������������ƿ����Һ�ܡ������ܺ͵ζ��ܵȣ���ʹ��ǰ�����뾭���У����ʹ��ʱ�����������ڱ���Ӧ��֤��ࡣ���ų�Һ��ʱ���������κ�ҺĤ�����ѡ�Ҫ������ȷ��ʹ�÷������в����Ͷ�����
4.1.3 ʹ��ĥ�ڲ�������ʱ��Ҫע��ĥ�ڵĹ���Ƿ�ȷ���ܷ�ʹ�ã�Ӧ��֤���ܲ�©����
4.1.4 ��ϴ��������ʱ�ɸ���������մ�������ѡ�÷�����ϴ�Ӽ������ϴҺ����ϴ����������ˮ��ϴ�ɾ��������ϴƿ��������ˮ��ϴ3��4�Σ����������ȱ�ʪ�������ˮ��Ϊ��
4.1.5 ʹ�ò�������ʱ��Ҫ������������(����������Ĵ�С�������ѹ����)ѡ�þ��к��ʿ����˰塣
4.2 ���������ʹ��
ȼ�������ҳ��õĴ���Ʒ�й�����������ʹ�õ����������������ۡ�����ȼ�Ϲܵȣ����Ǿ������õĿ�ʴ�Ժ������ԡ�����ʹ��ʱ��Ҫע����⼱����¶ȱ仯���������ѡ����������ڸ���������������������ã������������ڷֽ���Ʒ��
4.3 ʯӢ�����ʹ��
ȼ�������ҳ��õ�ʯӢ������Ҫ��ȼ�չܡ�ȼ����ȡ������������õ�������(�۵�1700��)�������ܹ����ܾ��ҵ��¶ȱ仯������1100��1200�濪ʼ��Ϊ����������ʧ����еǿ�ȡ����ʹ��ʯӢ����ʱӦ����������¶����¡�
4.4 ���������������ʹ�á�ά������ϴ
ȼ��������ʹ�õIJ���Ʒ�ܶ࣬��������������ȼ���ۡ��缫����˿�ȡ�������(��������������)���ŵ������£��ڸ����²����������ᡢ����ʴ����(������)����������������ú����������ⶨ������ʱ���ò���ȼ����ȼ��ú�������д�ȼ�յ����ܣ�ú������ȼ����ȫ����ʹ�ò���Ʒ(������������Ʒ)Ҫ��ѭһ������(�ɲμ��й�ȼ�������̻������ѧ�ֲ�)��
5 ��������ˮ���Լ�����Һ����������
�����ҹ����������õ�ˮ���Լ��Լ���Һ����������������鷽����Ҫ������Ա�������˽⡢ѡ��Ϳ���ˮ���Լ��Լ���Һ(��������Һ)��������
5.1 ��������ˮ����������
��������ˮ������ˮ������ˮ��ȥ����ˮ�ȡ�����ˮ������ϴ���������ȴ����ʹ�á�����ˮ����һ�������Ŀ���Լ����ƣ���ֵ�ⶨ�����Ƚ��ʺ�ϴ������ȣ�����ͨ����ˮ����CO2���ӷ����ᡢ�����������ȡ�������ķ�����Ŀʱ����ȥ����ˮ��ȥ����ˮ�ǽ�����ˮͨ�����������ӽ�����֬�����õ��Ĵ��Ƚϸߵ�ˮ��
����ˮ�Ĵ��ȿ��õ絼����ѧ�����顣������絼��ӦС��10��S/cm��pHֵӦΪ6.5��7.5����������������1.0mg/L�����������ɫ����ʱ������Ӧ����10min��
5.2 �Լ�����Һ����������
5.2.1 ������ѧ�Լ��Ĵ���(���)��Ϊ�ļ����ɸ��ݷ����ľ���Ҫ��ѡ�ã�
һ�� ��֤�Լ�(���GR) ����������
���� ������(���AR) ����һ�������Ҫ��ϸߵķ���
���� ��ѧ��(���CP) ����Ҫ��ϵ͵ķ���
�ļ� �����Լ�(���LR) ���Ƚϵͣ���������ʹ��
ѡ��ʱҪ�˶��Լ���ǩ�����в����ﺬ���Ƿ���Ϸ�����Ҫ�������������
�ڸ����Լ��ı����У���Ҫ��մ�ۡ������ʣ�ʹ�ô��Ȳ�������ʵ��Լ�����ʹ�������������
5.2.2 Ϊ��߷���������������������Լ�������ϵͳ���ɲ��ÿհ�����(�Լ��հͷ����հ�)�ķ�������У����
5.2.3 ���Լ��д��ڵĸ������ʣ������ؽᾧ������������ˮ�����ʵ��ܼ���ϴ�ķ��������Լ����������Լ������ʵĸ������á�
5.2.4 ���Ʊ���Һ������ˮ������Ҫ���߰���������ѡ���ܼ�������ʱҪ�ϸ������Ʊ���Һ��Ҫ����в��������Լ��ĸ���������ܽ⡢ϡ�͡���ϡ����ݡ�ת�Ƶȡ����ƺõı���ҺӦ������ĥ��Ӳ�ʲ����Լ�ƿ�У�ƿ��Ӧ�����Լ���ѧʽ��Ũ�ȡ������������������ڡ��Լ���������ߵı�ǩ�����ܹ���Ӱ������ߣ���������ɫƿ�С����Լ�ƿ��ȡ��Һʱ��ע�ⲻҪմ�۱�ǩ�����ӣ��Ѵ�ƿ��ȡ�����Լ������Բ������ٷ���ԭ��ƿ�С��������ʲ��ȶ��ı���Һ����Ҫ�����涨��ʱ�䱣�档
5.2.5 Ϊ��֤�Լ�����������ֹ�ڱ����ڼ���ʣ�����ˮ���⣬��飬���ն�����̼��Ϊ̼���Σ��ܸ��·ֽ⣬�ܹ��ձ��ʵȣ�Ӧ�����Լ������ԣ���ȷ��ѡ�ñ��ܡ������Լ��ķ��������ȡ�ܷ⡢�ܹ⡢���µȴ�ʩ�������ڱ��ܵ��Լ���ʹ��ʱ����ϸ��飬�ϸ����ʹ�á��������ʲ��ȶ����Լ���Ӧע���䱣�����Ч�ڻ��������(�ɴ������Ͽ�����������Ϊ8303254����Ϊ83��3��25�յ�4 ��)��
5.2.6 ������ȼ���ױ����綾�Լ�Ҫ��������־������ר�����Ʊ��ܡ���ȼ���ױ��Լ�Ӧ���������ͨ��ĵط����綾�Լ�Ӧ������ţ�����ʹ�ã����˹�ͬ�������Ǽ�������
5.2.7 ȡ���Լ����������ֿ��������Լ����ù���һ������ʹ���л��ܼ��ͻӷ���ǿ���Լ���Ӧ��ͨ����ڽ��в��������Բ�����ʹ������ֱ�Ӽ����л��ܼ�����Ũ�Ũ�ᡢǿ��������ʹ�ã�Ӧ���չ涨��Ҫ����в�����
6 ��Ʒ����������
6.1 ȼú�IJ���������
ȼú��һ����ɼ������ȵ��л����ң����Ѳɵ���������Ʒ��Ϊ�ˣ�����ʱ�����ϸ���ѭһ���IJ��������Ʊ���������ʱ��ҲҪ����һ���ij���涨��
6.1.1 ȼú�IJ����������ɰ���ԭˮ�粿��������������糧ȼ�����鷽������RS-1-1��83��RS-3-1��83�Ĺ涨ִ�С�
6.1.2 �����˹���������������Ϊ��������Ӧ������ʹ�û�е����װ�ò�������¯ú�Ļ�е����װ�õ����ܣ�Ӧ���ϡ���糧��¯ú��е����װ�ü��������й涨�ļ�������
6.2 ȼ�ͺ�����ȼ�ϵIJ�����
6.2.1 ȼ�͵IJ������ɰ���ԭˮ�粿��������������糧ȼ�����鷽������RS-28-1��83�Ĺ涨ִ�С�
6.2.2 ����ȼ�ϲ���Ŀǰ���ȶ��ķ������ѭ�����ɲο��й�ר��(��ˮ������������1984�����ġ�����ȼ�Ϸ�����)��
6.3 ��Ʒ�ı���
6.3.1 ȼú�ڿ���������������(ˮ�֡��ҷ֡��ӷ��ֺͷ����������仯)�������������ú�֣����ú�����������ʶ�ʧȥ�����ԡ�Ϊ�ˣ���ȼú��Ʒ�����ͺͱ����ڼ䣬Ӧ�����������������ʡ�
6.3.2 ȼú��Ʒ��÷�������ܷ�ǵ�����������Ӳ�����������ڣ�������Ӧ�������������������ǶԲⶨȫˮ�ֵ�ú������Ӧ�ܷ��װ���Ӳ���������ʱ�䲻����2d����������������ú��ƶú�������������ϴ���װ�������������ҽ��з�����ʱ�䣬Ҳ����̫����
6.3.3 ���������ķ�ú��Ʒ���Է������ĥ�����IJ���ƿ�����ܷ�ǵ�����ƿ���л�����ƿ�ڡ�����ú�����ڱ����������������һ�㱣����Ϊ3��6���¡�
6.3.4 ��Ҫ���ڱ����ú���������ڴ�ŵ������ڳ��뵪�������ܷ�ڣ�������������������������ʣ��羡����װ�������������������������������ھ�������������ˮ�еȡ�
7 �������ݵ���������
ʵ�з��������������ƣ������������������������õ���֤��ǰ���²�����Ч��������������������ָ��������ľ��ܶȺ�ȷ�ȵĿ��ơ�
7.1 ���ܶȵĿ���
���ܶ��DZ���������ݷ�ɢ�̶ȵ�һ��ָ�꣬��ƫ��ֵ(���βⶨֵ�����βⶨֵ��ƽ��ֵ�IJ�)�����ȡ����ܶ��ַ��ظ����ܶȺ����־��ܶȡ��ظ����ܶ�ָͬһ��������ͬһ�������������ݵľ��ܶȣ����־��ܶ���ָ��ͬ�������ڲ�ͬ�����ҡ���ͬ�������������ݵľ��ܶȡ�
�����ҿ��ƾ��ܶȵķ�����������Ϳ���ͼ����
7.1.1 ����� �������Ϊ�����ⶨֵ֮��(���βⶨ�����ֵ����Сֵ֮��)�����Ž�ֵ(ͨ����95%�����Ÿ�����)�����ǽ���ָ���ķ���������ָ���ı�����Ũ�ȶ��涨�ģ���ͬ�ķ��������ͱ���ɷֺ�����ͬʱ��������Ҳ����ͬ���������Ǿ����(Ӧ����8��)������Эͬ���飬ȡ�ô������ݣ��پ���ͳ�ƴ�����ȷ���Ĺ涨ֵ��������������������(����ΪT)���Ҽ�������(����ΪY)��ǰ�߿������ݵ��ظ����ܶȣ����߿������־��ܶȡ�
7.1.1.1 ���������� �����ڼ������������ظ����ܶȡ�����ƽ�����βⶨʱ�����βⶨֵ(x1��x2)�IJ�ֵδ��������������(����x1-x2����T)������Ϊ�����βⶨ�ľ��ܶ��ѴﵽҪ��ȡ����ƽ��ֵ��Ϊ���ս�������ͽг��˵������������һ�����ݿ��ɣ�Ҫ���е����βⶨ�������βⶨֵ�ļ���(�����βⶨֵ�е����ֵxmax����Сֵxmin�IJ�ֵ)С�ڶ��βⶨ�������1.2��ʱ(��xmax-xmin��1.2T)����ȡ�������ⶨֵ��ƽ��ֵ��Ϊ���������������Ҫ���е��Ĵβⶨ�����Ĵβⶨֵ�ļ���С�ڶ��βⶨ�������1.3��ʱ(���Ĵβⶨֵ�е�xmax-xmin��1.3T)����ȡ���ĸ��ⶨֵ��ƽ��ֵ��Ϊ�ⶨ��������˼�������������1.3�����������������ⶨֵ�ļ������������1.2��֮��ʱ�����ȡ�������ⶨֵ������ƽ��ֵ��Ϊ�ⶨ�������һ���������ȥ��������������δ�ﵽ����Ӧ��ȥȫ���ⶨ�����������������Լ��������ȣ��ҳ�ԭ����������²ⶨ��
7.1.1.2
�Ҽ�������
�����ڼ���������֮���ͬһ�����ⶨ����ľ��ܶ��Ƿ�ﵽҪ�������������Ҳⶨ�����ƽ��ֵ�IJ�ֵδ�����涨���Ҽ�������(����![]() ����Y)����Ϊ���������Ҽ�ⶨ�ľ��ܶȴﵽҪ���ͽг��˵������������һ��ƽ��ֵ���ɣ���������һ���������ڴ����нϴ������ʱ��Ӧͨ����ú���ķ������ݣ�ȷ�����ϴ��������(��6.2.1��6.2.2)��
����Y)����Ϊ���������Ҽ�ⶨ�ľ��ܶȴﵽҪ���ͽг��˵������������һ��ƽ��ֵ���ɣ���������һ���������ڴ����нϴ������ʱ��Ӧͨ����ú���ķ������ݣ�ȷ�����ϴ��������(��6.2.1��6.2.2)��
7.1.2
����ͼ�� ���Կ��ƾ��ܶȵĿ���ͼ������ͬһ����������һ������ú��(����ú��������ѡ��������ɻ�������������ú�������)��ͬ�������������£��ڲ�ͬʱ���ڷ���������20�Σ��������쳣ֵ�ķ���(����¼A�е�A2.1��A2.2)��ȥ�쳣ֵ�����µIJⶨֵӦ������20������������������貹��ⶨ���Բ���20�����ݡ�Ȼ�������ƽ��ֵ�ͱ���S(���㷽������¼�е�A2.1)���ټ������¿�����![]() ��3S�����¾�����
��3S�����¾�����![]() ��2S����Ϊ������CLƽ���ں����껭�����ܶȿ���ͼ(��ͼ1)��ͼ��UAL��LALΪ���¸����ߣ��ֱ�λ�ڡ�S����UWL��LWLΪ���¾����ߣ�����2S����UCL��LCLΪ���¿����ߣ�����3S����������Ϊ��ͬʱ���ڲⶨֵ��˳��n��
��2S����Ϊ������CLƽ���ں����껭�����ܶȿ���ͼ(��ͼ1)��ͼ��UAL��LALΪ���¸����ߣ��ֱ�λ�ڡ�S����UWL��LWLΪ���¾����ߣ�����2S����UCL��LCLΪ���¿����ߣ�����3S����������Ϊ��ͬʱ���ڲⶨֵ��˳��n��
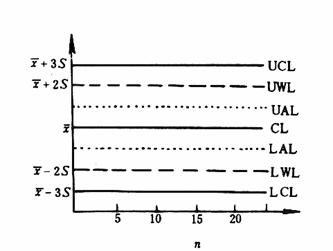
ͼ1 ���ܶȿ���ͼ
�ڷ�����������ͬʱ��Ҳ������������ֻ�ǶԿ������ķ���Ƶ�����ɸ��������ҵľ�������ʵ��������١����ڲ�������������Ա������������Ŀ�������ӶԿ������ķ���Ƶ�������ճ�������Ŀ�ķ�����һ��涨Ϊ20:1��������20��δ֪��ʱ���ɲ������һ����������������Ҳ����ѡ����֪��ֵ(�������ֵ)�ı�ú������ʱ�����ܿ�������ľ��ܶȣ�ͬʱҲ�ܿ���ȷ��(��7.2)��
���������ķ������(x)��ͼ�ڴ�㲢���������������������˽���������IJ���������Ա�������������п��ơ������ֵ��λ�ó������¾�����ʱ����Ӧ����ע�⣬��ʱ������������Ƿ�ʧ�ء������λ�ó������¿�����ʱ�����ߵ��������ߵ�һ���������ֻ�ӽ���������γ���ʱ�������ڲ���������Ӧֹͣ���������ʧ��ԭ��ֱ��������Խ�����ټ�������δ֪����
7.2 ȷ�ȵĿ���
ȷ���DZ��������������ʵֵ���ϳ̶ȵ�һ��ָ�꣬����˵�����ݵ�ȷ�ɿ��ԡ�ȷ�ȳ������ֵ�����ȣ����ֵ���ⶨֵ����ʵֵ֮����ֵ��������������ȷ����֮�����ֵ��С���������ݾ���ȷ����ʵֵ���ñ����ı�ֵ(�������ֵ)�����趨���������ı�ֵ�����������ʵ����(������8��ʵ����)��ͬ�ñ���������(���ܶ���ȷ�ȽԸߵķ�������)����Э�����飬ȡ�ô������ݾ�����ͳ�ƴ�����ȷ���ġ�ʹ�ñ�������ȷ�ȵķ��� ���£�
7.2.1 �ñ�ú���ı�ֵ����ȷ�� ��ú���ı�ֵ����һ��ȷ������ֵ������Ϊһ����ֵ�����䣬���̡���x����x��Ϊ��ֵ�IJ�ȷ���ȣ�������ʽ����ó���
����x=t����fS��
����ʽ���Կ�����ȷ���Ȧ�x��ⶨ������ƫ��(�ñ���S����ʾ)�йأ�ͬʱҲ�����ϵ��(������ƫ��ļ���)t�йء�ʽ��t����f��ֵ�ɲ�t�ٽ�ֵ�������ɲ�������f����(����A3)������Ϊ������ˮƽ����������ƫ��ֵ����S���ļ��ʣ���������ѡ����ͨ��ѡ��=0.05����ƫ�����S���ļ���Ϊ5%����f�����ɶȣ�f=m-1��mΪ�μ�Э���������������Ŀ��S����Ϊ��Э�������ҶԱ�����������ܱ����ú���Ħ̡�S����m���ɱ�ú����ʹ��˵�����ṩ��
�����ҶԱ�ú���ķ������x�����ڦ̡���x�����䣬��
![]()
����Ϊ���������ȷ�ģ���ƫ��̡���x����ȷ��ƫ����Զ������ȷ��������ϵͳ�����ڡ�
7.2.2 �ñ�ú�������������ȷ�� ��ú����������(����ΪB)Ҳ�������������Ҿ�Э�������õ�һ���涨ֵ����һ�����ҶԱ���Ʒ�ĵ��βⶨֵx��(��ƽ�м��βⶨ��ƽ��ֵx��)���ú���ı�ֵ��֮��δ����������ʱ����
![]()
����Ϊ�������ȷ������ͽг��˵�����������ȷ��������������ϵͳ�����ڡ�
���βⶨ�涨��������ֵ���βⶨƽ��ֵ�涨��������ֵ��ͬ�����ɱ�ú����ʹ��˵�����ṩ��
8 �������ݵĴ��������鱨��
8.1 �������ݴ���
���������ս���������ݱ���ģ��������ݵ�ȫ�����ֶ���������ʵ�ġ��Ӳ�����������ȫ�����й����ݵĴ�������������һ���ķ���(�й����ݴ����μ���¼��)��
8.2 ���鱨��
ȼ��������Ӧע������������ݣ�����ԭʼ�����¼����Ҫ�����������鱨�档�������ݵ���Чλ�������������Ĺ涨��д(��ԭˮ�粿��������������糧ȼ�����鷽������һ��涨)���Ĵ�������ʱ��Ӧ����ԭ�����������ּ����������˵�ǩ�֡���ȷ֪�ڷ����������й�ʧʱ������Ӧ���ϡ�������Ӧ��ԭʼ��¼�����鱨���Լ������������ϵ����������������ճ�������Χ�У����ֹ��������鹤��ͬ����Ҫ��
8.2.1 ��ԭʼ��¼��ԭʼ��¼��Ӧ������
��Ŀ���ƣ���Ʒ��ż���Ʒ�������������ڡ���������������йز������¶ȡ�ѹ��������ȡ���ҺŨ�ȡ�У��ֵ���հ�ֵ�ȣ�ȫ���������ݣ�����ʽ�����������������Ƶ������¼�����������������¼��������Աǩ�֣�������
ԭʼ��¼�ɽ������з���ʹ�õĸ���������涨�ļ��㹫ʽ�ֱ�һ��˳����ӡ���Ա�������Ա��¼�������ڲ��ġ�
8.2.2 ���鱨��ɲ��û�ҳ������װ������ʽ����������Ŀ����Ʒ��š��������ڡ����Խ����������Ҫ��˵����һ��˳���ӡ�����Խ�����뾭�ڶ��ߺ˸�����ʼ����д����Ӧ�����顢У�˼�������Ա��ǩ��(�����)������Ч��
8.2.3 ���ճ��ල�����ԭʼ��¼�ɱ������꣬�����м�ֵ�����鱨�棬���ݾ�������ʵ��ӳ������ڡ�
9 ������Ա�ļ�����ѵ�뿼��
ȼ�������ҵķ�������Ҫ�������뼼�����ϣ���Ҫ�߱�һ������֪ʶ��ʵ�ʲ������ܵ���Աȥִ�С�Ϊ�ˣ�������ѵ���˷�����Ա������֪ʶˮƽ�Ͳ��������������̶��Ƿdz���Ҫ�ġ�
9.1 ��ѵ
9.1.1 ��ѵ���� ÿһ�������ҵķ�����Ա���밴�似��������ܾ�����֮��Ӧ�Ļ�������֪ʶ��ʵ���������ѵ��������Ϊ��
�����������棺��������Ʒ��������������������������������ʹ�ú�У��������������ʹ�á�У����ά�ޣ���ȫ�����ȡ�
����֪ʶ������ԭ�������ݴ��������������Լ��������ϵIJ��������ȡ�
9.1.2 ��ѵ��ʽ ��ѵ�����ɲ�ȡ���ַ�ʽ������λ���ݾ���������������õķ�ʽΪ���ٰ������ѵ������й�רҵѧУ�������ޣ��ɼ�������ϸߵ���Ա����ָ���ʹ��ڲ��Լ��ܣ��ٰ�����Եļ���������ȡ�
9.2 ����
�������ܹ��ṩ�ϸ�ķ������ݣ��������֤������һΪ�ϸ�������豸����Ϊ�ϸ�ķ�����Ա����������Ϊ��Ҫ��Ϊ�ˣ�Ӧ���������������ĸ��ӳ̶��ɲ�ͬ�����������Աȥִ�У���ͬ�����������Ӧ������Ӧ������֪ʶ�Ͳ������ܡ����ڵضԷ�����Ա���п��ˣ������似�������Ա㰲��������ܹ�ʤ�εķ������������ֿ���Ҫ���й���Ա��ɵĿ���ίԱ��ȥִ�С�����֪ʶ��ͨ�����ԡ����Ի�������ʽ���飻�������ܿ�ͨ���ⶨ����Ʒ(�Ա�������Ա��˵������δ֪��)�ķ�ʽ�������������������������顣�������ҵ��ϼ������о���Ӧ���ڶ������ĸ������ҷ��������ú������ͳһ���ˣ�����������ҵĹ��� ������
�� ¼ A
�������ݵĴ�������
(�� �� ��)
A1 ������
���������е���Ч������ָ�÷�������ʵ���ܲ�����֣���������ʾ�����ֵ�Ĵ�С�����ұ�ʾ������ȷ�ȡ����յķ�������Ӧֻ������Ч���֣���Ч�����г�ȥĩλ���������⣬�������ֶ�����ʵ�ɿ��ġ�
A1.1 ������0��������
�����еġ�0������������Ч���֣�Ҳ����ֻ��λ���ö�������Ч���֣�Ӧ�Ӿ������������
(a)С�����ĩλ�ġ�0������Ч���֡�����Ϊ��ʾ��ȷ�����ˣ�9.8gӦдΪ9.800g������������0��������Ч�ġ�
(b)�ǡ�0������ǰ�ġ�0����������Ч���֡����磬0.0032g��ǰ������0����������Ч���֡�
(c)λ����Ч����֮��ġ�0��������Ч���֡�����0.503mg/L�У�5��3֮��ġ�0������Ч���֡�
(d)���������ġ�0����������Ч���֣�Ҳ���Բ��ǣ�������������ƽ(����Ϊ0.1g)��һ����֮����Ϊ1.5g�����ú��˱�ʾ����Ϊ1500mg����ʱ����ĩ��������0��������Ч���֣�����ø���Ϊ0.001g��ƽ�Ƶ���������֮����Ϊ1.500g������ʾΪ1500mgʱ������ĩ��������0��������Ч���֡���ˣ�������ġ�0���Ƿ�Ϊ��Ч�������Ӿ�������жϡ�Ϊ������ᣬ���ÿ�ѧ����������1500mg������ĩ������0��������Ч���֣���д��1.5��10-3g���������ø������ܶȵİ취���������д��1500��100mg���������ĩ������0��������Ч ���֡�
��Ч�����ܹ���ӳ��һ���ض���������ȷ�ȵķ�Χ��Ϊʹ���շ�����������������壬���Ϸ�����Ŀ�ģ���Ч�������ﵽ����λ�����ܳ���������ͼ��Ũ�ȵ���Ч�������ܴﵽ����λ�����������������б���С������λ����������ݵ�ȷ�ȡ�����Ҫ�������Ч����ʱ��Ӧ��һ�����Ʒ�������������ѡ���� ��������
A1.2 ��Ч���ֵ���Լ����
��Ӧ��������Ч������������֣�����������������˫����Լ����ִ��(�����ұ�GB 1.1-81��¼C��������Լ����)����������ǣ������������ֵ�һλ�����ڡ�5���������1����С�ڡ�5������ȥ���������ڡ�5��ʱ����ǰһλΪ�����������1����Ϊ˫������Ϊż��(������0��)������ȥ����������5�����滹�����ֲ���ȫΪ��0��ʱ�������1�������������������ֲ���һ������ʱ�������������ж����Լ��Ӧ�������涨�������ĵ�һ�����ֽ�����Լ��
����������������Լ��ֻ����һλС����
��Լǰ ��Լ��
14.2432 14.2
26.4843 26.5
1.0501 1.1
0.3500 0.4
1.0500 1.0
��13.4546��Լ��ֻ������λ��Ч����
����ȷ�������ǣ�13.4546��13.455��13.46��13.5��14��
��ȷ�������ǣ�13.4546��13��
A1.3 ��Ч��������ı�������
���Ӽ�������ʱ����������Ч���ֵı�����Ӧ�Բμ�����ĸ�����С�����λ�����ٵ���Ϊ�����˳�����ʱ����������Ч���ֵı�����Ӧ�Բμ�����ĸ�������Ч����λ�����ٵ���Ϊ���������Ӧ��������Ч������ֻ����һλ�������֡�
�Ӽ���ʾ����0.0121+25.64+1.05792
��ȷ�ļ�����Ӧ����С������λ����26.71������Ӧ��26.70992��
�˳���ʾ����0.0121��25.64��1.0578
��ȷ�ļ�����Ӧ������λ��Ч���֣���0.328����������������֡�
A1.4 ���ܶȵ���Ч����
��ʾ����������ܶȵ�����(��ƫ��ֵ�����͡���������)ֻȡһλ��Ч���֣������������ܶ�ʱ������ȡ��λ��Ч���֣������ֻ��ȡ��λ��
�ڼ����͡��������ʱ��Ϊ�˼��ټ����������ϼ���������Բ��ö���ʽ���õ�Чʽ���£�
����ʽ ��Чʽ
���
![]() (A1)
(A1)
�� ��
![]() (A2)
(A2)
����
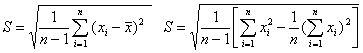 (A3)
(A3)
��ÿһ����������У����м����ݲ�����Լ��ֻ��Լ����������Ҫ���λ��(һλ����λ)��
A1.5 ƽ��ֵ����Ч����
�ڼ��㲻�����ĸ��ⶨֵ��ƽ��ֵʱ��ƽ��ֵ����Ч����λ������һλ��
�������� 3.77��3.70��3.79��3.80��3.72��ƽ��ֵ��
����
![]()
A1.6 �˷��Ϳ�������Ч����
�ⶨֵ�˷��Ϳ���ʱ��ԭ�ⶨֵ�м�λ��Ч���֣��������ͱ�����λ��Ч���֡�
��������6.542��![]() ��
��
6.542=42.7716 Ӧ������λ��Ч����Ϊ42.8
![]() Ӧ������λ��Ч����Ϊ2.72
Ӧ������λ��Ч����Ϊ2.72
A1.7 �����ͷ���������Ч����
�ⶨֵ�Ķ������㣬��ȡ����С������λ��(����������)����������Ч���ֵ�λ����ͬ��
�������H����Ϊ7.98��10-2mol/L��Һ��pH��
��H����=7.98��10-2mol/L
pH=-lg��H����=-lg(7.98��10-2)��1.098
��pHΪ3.20��Һ�ģ�H���ݣ�
pH=-lg��H����=3.20
��H����=6.3��10-4mol/L
A1.8 ����ʽ�е���ѧ�����������������ȵ���Ч����
�����м���ʽ�е���ѧ�����С�e��ijЩ����������������������������ֵ���Լ�������������ȫ�������ۼ������õ�����ֵ������Ч���ֵ�λ������Ϊ���ޡ�������ֵ�ڼ�������Ҫ��λ�Ϳ���д��λ�����磬�����������ʽ![]() �е�
�е�![]() ��1m=100cm�е�100���ⶨ����n�����ɶ�f��H2SO4�е�2��4���Լ�K2Cr2O7��������ԭ��Ӧ��Ħ�������Ļ�����Ԫ
��1m=100cm�е�100���ⶨ����n�����ɶ�f��H2SO4�е�2��4���Լ�K2Cr2O7��������ԭ��Ӧ��Ħ�������Ļ�����Ԫ![]() �е�
�е�![]() ��
��
A2 ������ֵ��������
��һ��ⶨֵ(����Ӧ���������������ϵ���ֵ)�г���һ����ƫ��ƽ��ֵ��Զ�Ŀ�����ֵ�����ⶨֵ��Ŀ����ʱ��������������Щ��ֵ�Լ���ƽ��ֵ������ϴ��Ӱ�졣Ϊ�ж���Щ������ֵ�Ƿ�Ӧ��������������Ӧ����һ����ͳ�Ʒ����м��飬�Ծ������䱣������������
A2.1 Grubbs���鷨
�����������ݰ��������ֲ�ͬ���������
(a)ֻ��һ��������ֵʱ�����ⶨֵ����С����˳�����У�x1��x2��x3����xn���������й�ʽ���ͳ����G1��Gn��
��![]() (A4)
(A4)
ʽ�� ![]() Ϊn���ⶨֵ��ƽ��ֵ������
Ϊn���ⶨֵ��ƽ��ֵ������
![]() (A5)
(A5)
��SΪn���ⶨֵ�ı����
![]() (A6)
(A6)
�ɱ�A4�����Ӧ�ڲⶨ����n��������ˮƽ�����ٽ�ֵGa��n����Ga��n����G1��Gn����Gn��Ga��n������ȥxn����G1��Ga��n������ȥx1��
(b)�������������������ڵĿ�������ʱ������Ӧ�����ڲ��һ�����ݣ�����x1��x2��ͬ��������ֵʱ����ͨ������G2����x2�Ƿ�Ӧ����������x2����������x1ҲӦ������ֻ���ڼ���x2ʱ���ⶨ����Ӧ�ټ�һ�Ρ�
(c)����������������λ��ƽ��ֵ����Ŀ�����ֵʱ��Ӧ�ֱ������������ֵ������һ�����ݾ�����ȥ�����ڼ�����һ����ֵʱ�����ⶨ�����ټ���һ���⣬��Ӧ��ѡ99%������ˮƽ(����=0.01)��
������������һ����С�������еIJⶨֵ����ȥ���е��쳣ֵ��
40.02��40.12��40.16��40.18��40.20��
����n=6��![]() ��S=0.067������ͳ����
��S=0.067������ͳ����
![]()
���A4��ѡ��=0.05����G0.05=1.82����G1��G0.05�����Դ�ֵ���Ա�����
A2.2 Q(Dixon)�����鷨
���ⶨֵ��С����˳�����У�x1��x2��x3����xn������x1��xnΪ������ֵ����������ֵ�����ڽ���ֵ�IJ�ֵ������ȫ����ֵ�ļ���ó�Qֵ��
![]() (A7)
(A7)
���������Qֵ�����ɱ�A5�в�����ٽ�ֵQ0.05ʱ�����ֵΪ�쳣ֵӦ��ȥ����Q0.05��Q��Q0.01�����ֵΪƫ��ֵ����Q��Q0.05�����ֵΪ����ֵ�����ⶨֵ��8������ʱ������Qֵ�Ĺ�ʽ���ڱ�A5�С�
������������һ����С�������еIJⶨֵ���������е��쳣ֵ��14.65��14.90��14.90��14.92��14.95��14.96��15.00��15.00��15.01��15.02��
����n=10����x1����ʱ������A5�еĹ�ʽ����Q��
![]()
���A5��ѡ��=0.05����Q0.05=0.477����Q��Q0.05������14.90Ӧ��ȥ��
A3 �����ߵĻ���
���Բⶨ��Ŀ��Ҫ��������ʱ��Ӧ��ֱ�ع鷨�Ա����߽���У��������ʵ�ʷ��������д����Ÿ���������أ�ʵ��ĸ�����㣬����ȫ����һ��ֱ���ϣ����ûع鷨��������Ը�������������С��ֱ�߷���ʽ�����ô�ֱ�߷���ʽ��������x��y���룬���ɻ��Ƴ�һ�������ߡ�
��xΪ�Ա�����yΪ�������ֱ�߷���ʽΪ
![]() ��(A8)
��(A8)
ʽ�У�bΪֱ�ߵ�б�ʣ��蹲�ⶨn�Σ���
![]() (A9)
(A9)
aΪֱ����x���ϵĽؾ࣬��
![]() (A0)
(A0)
��������֪�����ʵĺ���Ϊx��������Ӧ����Ϊy������x2��y2��xy�����ܺ����ڱ�A1�С�
�����е�ֵ����a��b��r�Ĺ�ʽ�У�ʽ��n=6����
![]()
![]()
�õ���ֱ�߷���Ϊ
![]()
�� A1
|
�ⶨ����n |
x |
y |
x2 |
y2 |
xy |
|
1 |
1.00 |
0.079 |
1.00 |
0.0062 |
0.079 |
|
2 |
2.00 |
0.142 |
4.00 |
0.0202 |
0.284 |
|
3 |
4.00 |
0.317 |
16.0 |
0.100 |
1.27 |
|
4 |
6.00 |
0.480 |
36.0 |
0.230 |
2.88 |
|
5 |
8.00 |
0.633 |
64.0 |
0.401 |
5.06 |
|
6 |
10.00 |
0.800 |
100.0 |
0.640 |
8.00 |
|
�� |
31.00 |
2.451 |
221.0 |
1.397 |
17.58 |
�ڻ�ͼʱ��xֵ����ѡ�����������糣ѡ��0��2.00��4.00����������ֱ�߷���ʽ����y����Ӧֵ��
![]()
![]()
![]()
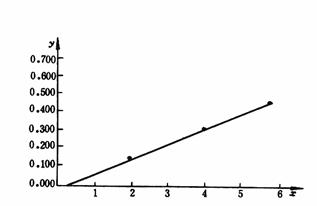
ͼA1 ֱ�ع鷨��ͼ
��������������ֵ�㣬��������ֽ�ϣ����ܻ��һ��ֱ�ߣ�����ֱ�߶�����ʵ���������˵���������С��һ��ֱ�ߣ���ͼA1��ʾ��
�����ع�ֱ����ʱ����ϸ��ӣ�Ϊ��߹���Ч�ʣ�������Ա���뱸��ͳ�Ƽ��㹦�ܵļ������������������������������ѡ�ü�����ʱ�����ѡ�ð����лع����������ܼ������͡�ʹ�����ּ��������ڽ��б����ʵĺ�����ⶨֵ�Ļع����ʱ��ֻҪ����������ʵĸ��ֺ������Ӧ�IJⶨֵ����������ڣ�������ó�����a���ع�ϵ��b��ͬʱ�ڲⶨδ֪����ʱ��ֻҪ��δ֪�����IJⶨֵ����������ڣ��������δ֪�����ĺ���ֵ�������ü�������ͳ�ƺͻع����ľ�����������ɼ���������ʹ��˵���顣
A4 ���õ�ͳ�Ƽ��㷽��
��ѧ�����������������������ʵ��������Ӷ��������ʵ����Ժ���ɡ����ǣ���ѧ���������������ⶨֵͨ����������(�����㹫ʽ)��õģ�������ֱ�Ӳ��������ġ��������ⶨֵ�����ɱ�����ܵ����۸������ص�Ӱ�죬���л���С������������Щ����ֳ���������һ��ʹ��ѧ�����Ľ��ƫ������ֵ����ƫ��ij̶�Ҳ����һ�£�������䡣Ҫ�ֱ�����ƫ���Ƿ��˹涨���ޣ�ʹ���������Ϊ���ɿ�����ȷ����������ѧ������֪ʶ���ֶ��Dz����ģ�����Ҫ����������ͳ�Ƽ��鷽��������ֱ�С�IJ��죬�������Է�������ʱ��ʹ��ͳ�Ƽ��鷨��������Ч�ġ�
һ��������ϵ����������������������������һ����Ϊ��������ֵ����ƽ��ֵ����һ����Ϊ��ɢ����ֵ��������S��ֻҪ��֪��������������ֵ��һ��������ϵ�������Ϳ��Ա�������Ϊ�ˣ�����ƽ��ֵ�ͱ����Ƿ�ƫ������ֵ��ͳ�Ƽ���ĺ������⡣����ʵ�ʹ����в�õ���������ֵ��ƫ����ֵ�Dz����ܵģ��ؼ���ȷ������ƫ��ij̶ȡ���ƫ�볬��һ������(��ͳ�Ƽ����г��ٽ�ֵ)������Ϊ�ⶨֵ����ֵ���������Բ��죬��õĽ����ȷ����ȼ�ϼ��鹤���г��õ�ͳ�Ƽ��鷨��F���鷨��t���鷨����
A4.1 F���鷨
A4.1.1F���鷨��Ӧ�� F���鷨�����ڼ��鱻����ϵ�ķ�ɢ�̶�(��ɢ��)����һ��ⶨֵ��˵�������Ǽ����侫�ܶȵģ�����֮�����Ǽ��鷽��S2��һ�ַ���������������������Ʒ�Ƿ����һ�£�����ú����ԭú�������Ƿ�һ�£���������Ƿ�ﵽ�˹涨�ľ��ܶ�ֵ�ȡ�
A4.1.2
F����ij���
�������������ϵ�ķ���![]() (������¼A1.4�м��㷽��Ĺ�ʽ)�������������ϵ�ķ���
(������¼A1.4�м��㷽��Ĺ�ʽ)�������������ϵ�ķ���![]() ���������������ʽ�����ֵF��
���������������ʽ�����ֵF��
![]() (A11)
(A11)
ʽ�з��ӵķ���ֵҪ���ڷ�ĸ�ķ���ֵ����ʹ��ֵF����1������������FֵС���ɱ�A6(F�ٽ�ֵ��)������ٽ�ֵ![]() ������Ϊ���������������Բ��졣��֮����Ϊ���������������Բ��졣�ɱ�A6���ٽ�ֵ
������Ϊ���������������Բ��졣��֮����Ϊ���������������Բ��졣�ɱ�A6���ٽ�ֵ![]() ʱ��Ҫ�����������ɶȣ���һ���ɶ�f1=n1-1(n1Ϊ��IJⶨ����)�͵ڶ����ɶ�f2=n2-1(n2ΪС����IJⶨ����)��ͬʱҲҪѡһ���ʵ���������ˮƽ��(ͨ��ѡ��=0.05)����A6Ϊ����F�ٽ�ֵ����Ϊ�ˣ��ڲ��ʱ��Ҫע�������������һ������Dz���֪��������������ߴ�ֻҪ�����������Բ�����ˣ���ʱӦ����˫���飬�����A6ʱ����ѡ���Ħ�ֵ����
ʱ��Ҫ�����������ɶȣ���һ���ɶ�f1=n1-1(n1Ϊ��IJⶨ����)�͵ڶ����ɶ�f2=n2-1(n2ΪС����IJⶨ����)��ͬʱҲҪѡһ���ʵ���������ˮƽ��(ͨ��ѡ��=0.05)����A6Ϊ����F�ٽ�ֵ����Ϊ�ˣ��ڲ��ʱ��Ҫע�������������һ������Dz���֪��������������ߴ�ֻҪ�����������Բ�����ˣ���ʱӦ����˫���飬�����A6ʱ����ѡ���Ħ�ֵ����![]() ��Ȼ����
��Ȼ����![]() ���ڶ��������Ҫȷ�����������е�һ������������һ������ʱӦ���õ����飬���ʱ
���ظı��ֵ��
���ڶ��������Ҫȷ�����������е�һ������������һ������ʱӦ���õ����飬���ʱ
���ظı��ֵ��
���������ֲ�ú���ķ�������ͬһ��ú�и���16����������ûҷֵĽ�����A2���У��ж������ֲ����������������Բ��졣
��A2 ���ֲ����������������Ļҷ�(Ag)
|
�� �� �� |
��������һ |
���������� |
�� �� �� |
��������һ |
���������� |
|
1 2 3 4 5 6 7 8 |
20.74 25.41 30.07 26.45 24.91 25.06 30.44 29.98 |
22.15 28.25 28.60 30.00 26.90 27.20 31.80 29.30 |
9 10 11 12 13 14 15 16 |
29.44 24.60 24.93 28.13 27.93 27.61 30.87 29.47 |
29.05 29.50 29.30 27.90 23.80 28.85 30.00 29.20 |
�ӱ��п��Կ��������ݼ�Ϊ��ɢ����������ú�������������ȡ�����Ŀ����Ϊ���������ֲ��������Ƿ��������Բ�ͬ�����ǿ���ú�ʾ�������Ρ�Ϊ�ˣ�������������ݵķ���S2��Fֵ��
S21=7.90�� S22=5.62
![]()
��Ϊ����Ҫ֪�������Ǹ����������Ҫ��˫���顣��ѡ��=0.05�����ڲ��ʱ��![]() ���������ɶȸ�Ϊ16-1=15���ɱ�A6�в��F0.025��15��15=2.86������
���������ɶȸ�Ϊ16-1=15���ɱ�A6�в��F0.025��15��15=2.86������![]() ���������ֲ����������������ԵIJ�ͬ��
���������ֲ����������������ԵIJ�ͬ��
A4.2 t���鷨
A4.2.1 t���鷨��Ӧ�� t���鷨�����ڼ��鱻����ϵ�ļ��г̶�(����ֵ��ƽ��ֵ)��һ�ַ���������ƽ��ֵ����ֵ�ıȽϣ�����ƽ��ֵ�ıȽ��Լ���ͬ���������ıȽϵȡ�
A4.2.2 t����ij��� ����t����ʱ��Ӧ����F����(ƽ��ֵ����ֵ�Ƚ�ʱ�ɲ���)��֤��������ϵ�ķ����������Բ��������ƽ��ֵ��t���顣����ʱ������ʽ���tֵ��
![]() (A12)
(A12)
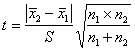 (A13)
(A13)
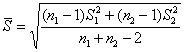 (A14)
(A14)
ʽ�� �̡�����ֵ��
S�����ⶨֵ�ı��
S21��S22������һ���ڶ�������ϵ�ⶨֵ�ķ��
![]() ������һ���ڶ�������ϵ�ⶨֵ��ƽ��ֵ��
������һ���ڶ�������ϵ�ⶨֵ��ƽ��ֵ��
![]() ������һ�͵ڶ�������ϵ�ⶨֵ��ƽ�����
������һ�͵ڶ�������ϵ�ⶨֵ��ƽ�����
n1��n2������һ���ڶ�������ϵ�IJⶨ������
�Ƚ�ƽ��ֵ����ֵʱ��ʽ(A12)���㣬�Ƚ�����ƽ��ֵʱ��ʽ(A13)���㡣
���������õ�tֵ����A3���õ�t�ٽ�ֵt��,f�Ƚϡ����ʱ��������ˮƽ�������ɶ�f��������������ѡ��һ��ѡ��=0.05��f=n-1����A3Ϊ˫�����������������ʱ���뽫������2(��2��)���ж����������Բ�������ݣ�
��t����t0.05 �������Բ���
t0.05����t����t0.01 �������Բ���
��t����t0.01 �зdz������Բ���
��������֪��ֵΪ18106J/g�ı�ú������һ̨�����ȼƣ�����6�Σ��������£�18073��18106��18136��18081��18115��18052��
�������ý�����ֵ��=18106J/g�Ƿ�һ�¡�
������ȷ���ⶨ��ƽ��ֵ���ֵ�ĸ�������Ӧ����˫���顣�����ƽ��ֵ=18094�ͱ���S=30.734��Ȼ�����
![]()
���A3���ٽ�ֵt��,f=t0.05,5=2.57���Ƚ�t��t��,f����t��t��,f�����ԣ�����õ�6��ƽ��ֵ������ı�ֵ�������Բ��졣
��A3 t�� �� ֵ �� (˫��)
|
�� |
f |
|||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
78 |
9 |
10 |
|
|
|
0.01 |
63.66 |
9.93 |
5.84 |
4.60 |
4.03 |
3.71 |
3.50 |
3.36 |
3.25 |
3.17 |
|
0.05 |
12.71 |
4.30 |
3.18 |
2.78 |
2.57 |
2.45 |
2.37 |
2.31 |
2.26 |
2.23 |
|
0.1 |
6.31 |
2.92 |
2.35 |
2.13 |
2.02 |
1.94 |
1.90 |
1.86 |
1.83 |
1.81 |
��A4 Grubbs �� �� ֵ �� (˫��)
|
�� |
n |
|||||||||
|
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
|
|
0.01 |
1.15 |
1.49 |
1.75 |
1.94 |
2.10 |
2.22 |
2.32 |
2.41 |
2.48 |
2.55 |
|
0.05 |
1.15 |
1.46 |
1.67 |
1.82 |
1.94 |
2.03 |
2.11 |
2.18 |
2.24 |
2.29 |
��A5 Q ֵ �� (Dixon�ٽ�ֵ)
|
�ⶨ����n |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
||
|
��=0.05 |
0.941 |
0.765 |
0.642 |
0.560 |
0.507 |
0.554 |
0.512 |
0.477 |
||
|
��=0.01 |
0.988 |
0.889 |
0.780 |
0.698 |
0.637 |
0.683 |
0.635 |
0.597 |
||
|
����x1ʱ |
|
|
||||||||
|
����xnʱ |
|
|
||||||||
|
�ⶨ����n |
11 |
12 |
13 |
14 |
15 |
16 |
17 |
18 |
||
|
��=0.05 |
0.576 |
0.546 |
0.521 |
0.546 |
0.525 |
0.507 |
0.490 |
0.475 |
||
|
��=0.01 |
0.679 |
0.642 |
0.615 |
0.641 |
0.616 |
0.595 |
0.577 |
0.561 |
||
|
����x1ʱ |
|
|||||||||
|
����xnʱ |
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
��������ú�ʼ���Ա��ͬһ������ͬһ������ͬһ�����ⶨ̼Ԫ�غ���������6�Σ��������£�
�� 91.16�� 90.34�� 90.58�� 90.89�� 91.06�� 90.46
�� 91.93�� 92.10�� 91.18�� 91.44�� 91.71�� 91.64
�ԱȽ϶����������Ƿ��������Բ�ͬ��
���ڲ���ȷ�������˵IJⶨ���˭��˭С��������˫���顣�����������ݵ�ƽ��ֵ�ͱ��
��![]() ��
S1=0.33��
n=6
��
S1=0.33��
n=6
��![]() ��
S2=0.33��
n=6
��
S2=0.33��
n=6
�������ݵı�����ͬ(S1=S2)��˵���ⶨ�ľ��ܶ���һ�µģ���ƽ��ֵȴ����ͬ����t���鷨�ж�����ƽ��ֵ�Ƿ����������졣�����������ݵı�����ͬ������ƽ��������Ϊ0.33������ʽ(A13)����tֵ
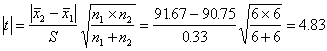
ѡ��=0.05�� f=n1+n2-2=6+6-2=10�����A3��t���ٽ�ֵt��=t0.05��10=2.23��t0.01��10=3.17���֣�t����t0.01��˵����������Ա�ķ�������зdz������IJ��졣�����ж���һ������Ա�ķ������ȷ����Ҫ����֪��ֵ�ı�ú�����з������������ж�(����������7.2)��
��A6 F �� �� ֵ �� (����) ��=0.025
|
f2 |
f1 |
|||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
12 |
15 |
|
|
1 |
648.0 |
800.0 |
864.0 |
900.0 |
922.0 |
937.0 |
948.0 |
957.0 |
963.0 |
969.0 |
976.7 |
984.9 |
|
2 |
38.5 |
39.0 |
39.2 |
39.2 |
39.3 |
39.3 |
39.4 |
39.4 |
39.4 |
39.4 |
39.41 |
39.43 |
|
3 |
17.4 |
16.0 |
15.4 |
15.1 |
14.9 |
14.7 |
14.6 |
14.5 |
14.5 |
14.4 |
14.34 |
14.25 |
|
4 |
12.2 |
10.6 |
9.98 |
9.60 |
9.36 |
9.20 |
9.07 |
8.98 |
8.90 |
8.84 |
8.75 |
8.66 |
|
5 |
10.0 |
8.43 |
7.66 |
7.39 |
1.15 |
6.98 |
6.85 |
6.76 |
6.68 |
6.62 |
6.52 |
6.43 |
|
6 |
8.81 |
7.26 |
6.60 |
6.23 |
5.99 |
5.82 |
5.70 |
5.60 |
5.52 |
5.46 |
5.37 |
5.27 |
|
7 |
8.07 |
6.54 |
5.89 |
5.52 |
5.29 |
5.12 |
4.99 |
4.90 |
4.82 |
4.76 |
4.67 |
4.57 |
|
8 |
7.57 |
6.06 |
5.42 |
5.05 |
4.82 |
4.65 |
4.53 |
4.43 |
4.36 |
4.30 |
4.20 |
4.10 |
|
9 |
7.21 |
5.71 |
5.08 |
4.72 |
4.48 |
4.32 |
4.20 |
4.10 |
4.03 |
3.96 |
3.87 |
3.77 |
|
10 |
6.94 |
5.46 |
4.83 |
4.47 |
4.24 |
4.07 |
3.95 |
3.85 |
3.78 |
3.72 |
3.62 |
3.52 |
|
12 |
6.55 |
5.10 |
4.47 |
4.12 |
3.89 |
3.73 |
3.61 |
3.51 |
3.44 |
3.37 |
3.28 |
3.18 |
|
15 |
6.20 |
4.77 |
4.15 |
3.80 |
3.58 |
3.41 |
3.29 |
3.20 |
3.12 |
3.06 |
2.96 |
2.86 |
��=0.05
|
f2 |
f1 |
|||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
12 |
15 |
|
|
1 |
161.0 |
200.0 |
216.0 |
225.0 |
230.0 |
234.0 |
237.0 |
239.0 |
241.0 |
242.0 |
|
|
|
2 |
18.5 |
19.0 |
19.2 |
19.2 |
19.3 |
19.3 |
19.4 |
19.4 |
19.4 |
19.4 |
|
|
|
3 |
10.1 |
9.55 |
9.28 |
9.12 |
9.01 |
8.94 |
8.89 |
8.85 |
8.81 |
8.79 |
|
|
|
4 |
7.71 |
6.94 |
6.59 |
6.39 |
6.26 |
6.16 |
6.09 |
6.04 |
6.00 |
5.96 |
|
|
|
5 |
6.61 |
5.79 |
5.41 |
5.19 |
5.05 |
4.95 |
4.88 |
4.82 |
4.77 |
4.74 |
|
|
|
6 |
5.99 |
5.14 |
4.76 |
4.53 |
4.39 |
4.28 |
4.21 |
4.15 |
4.10 |
4.06 |
|
|
|
7 |
5.59 |
4.74 |
4.35 |
4.12 |
3.97 |
3.87 |
3.79 |
3.73 |
3.68 |
3.64 |
|
|
|
8 |
5.32 |
4.46 |
4.07 |
3.84 |
3.69 |
3.58 |
3.50 |
3.44 |
3.39 |
3.35 |
|
|
|
9 |
5.12 |
4.26 |
3.86 |
3.63 |
3.48 |
3.37 |
3.29 |
3.23 |
3.18 |
3.14 |
|
|
|
10 |
4.96 |
4.10 |
3.71 |
3.48 |
3.33 |
3.22 |
3.14 |
3.07 |
3.02 |
2.98 |
|
|
______________________
����˵��:
������ݵ�λ����ˮ������ѧԺ��ɽ�����������о���������������ѧ�о�����
������Ҫ��������������ܳ��䡢�����١�